
ELAHERE 5 mg-mL, solution à diluer pour perfusion, boîte de 1 flacon de 20 ml
Dernière révision : 19/11/2024
Taux de TVA : 10%
Laboratoire exploitant : ABBVIE
Source :

ELAHERE en monothérapie est indiqué dans le traitement des patientes adultes présentant un cancer épithélial séreux de haut grade de l'ovaire, des trompes de Fallope ou péritonéal primitif, positif au récepteur alpha du folate (FRα), résistant aux sels de platine et qui ont reçu une à trois lignes de traitement systémique antérieures (voir rubrique Posologie et mode d'administration).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du médicament administré doivent être clairement enregistrés.
Affections oculaires
Le mirvétuximab soravtansine peut provoquer des effets indésirables oculaires sévères, incluant des troubles visuels (vision trouble principalement), une kératopathie (troubles cornéens), une sécheresse oculaire, une photophobie et une douleur oculaire (voir rubriques Effets sur l'aptitude à conduire des véhicules et à utiliser des machines et Effets indésirables).
Les patientes doivent être adressées à un ophtalmologue pour un examen ophtalmologique avant l'instauration du traitement par mirvétuximab soravtansine.
Avant le début de chaque cycle, il faut conseiller aux patientes de signaler au médecin prescripteur ou à un professionnel de santé qualifié tout nouveau symptôme oculaire ou toute aggravation d'un symptôme oculaire.
En cas d'apparition de symptômes oculaires, un examen ophtalmologique devra être réalisé, le compte rendu devra être revu et une modification de la dose de mirvétuximab soravtansine pourra être nécessaire en fonction de la sévérité des anomalies constatées (voir rubrique Posologie et mode d'administration).
L'utilisation de larmes artificielles pendant le traitement par mirvétuximab soravtansine est recommandée. Chez les patientes présentant des effets indésirables cornéens de grade ≥ 2, l'utilisation de corticoïdes topiques ophtalmiques est recommandée lors des cycles ultérieurs de mirvétuximab soravtansine (voir rubrique Posologie et mode d'administration).
Le médecin doit surveiller la patiente afin de détecter une toxicité oculaire et doit suspendre le traitement par mirvétuximab soravtansine, réduire la dose ou arrêter définitivement le traitement en fonction de la sévérité et de la persistance des effets indésirables oculaires (voir rubrique Posologie et mode d'administration).
Les patientes doivent être informées que, sauf avis contraire d'un médecin, elles doivent éviter de porter des lentilles de contact pendant le traitement par le mirvétuximab soravtansine.
Pneumopathie inflammatoire
Une pneumopathie interstitielle, y compris une pneumopathie inflammatoire, sévère, engageant le pronostic vital ou d'issue fatale, peut survenir chez les patientes traitées par le mirvétuximab soravtansine (voir rubrique Effets indésirables).
Les patientes doivent être surveillées afin de détecter l'apparition de signes et symptômes pulmonaires de pneumopathie inflammatoire, tels qu'une hypoxie, une toux, une dyspnée ou des infiltrats interstitiels à la radiographie. Les étiologies infectieuses, cancéreuses et les autres causes de ces symptômes doivent être exclues à l'aide d'investigations appropriées.
Chez les patientes qui présentent une pneumopathie inflammatoire de grade 2 persistante ou récidivante, le traitement par mirvétuximab soravtansine doit être suspendu jusqu'au retour des symptômes à un grade ≤ 1 et une réduction de dose doit être envisagée. Le traitement par mirvétuximab soravtansine doit être arrêté définitivement chez toutes les patientes présentant une pneumopathie inflammatoire de grade 3 ou 4 (voir rubrique Posologie et mode d'administration). Chez les patientes asymptomatiques, le traitement par mirvétuximab soravtansine peut être poursuivi avec une surveillance étroite.
Neuropathie périphérique
Des cas de neuropathie périphérique, incluant des réactions de grade ≥ 3, sont survenus avec le mirvétuximab soravtansine (voir rubrique Effets indésirables).
Les patientes doivent être surveillées afin de détecter l'apparition de signes et symptômes de neuropathie tels que paresthésie, sensations de fourmillements ou de brûlure, douleur neuropathique, faiblesse musculaire ou dysesthésie. Chez les patientes présentant une neuropathie périphérique nouvelle ou s'aggravant, le traitement par mirvétuximab soravtansine doit être suspendu, la dose réduite ou le traitement définitivement arrêté selon la sévérité de la neuropathie périphérique (voir rubrique Posologie et mode d'administration).
Toxicité embryonnaire et fœtale
Sur la base de son mécanisme d'action, le mirvétuximab soravtansine lorsqu'il est administré à une patiente enceinte, peut avoir des effets délétères sur l'embryon ou le fœtus car il contient un composé génotoxique (DM4) qui agit sur les cellules en division.
Les patientes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par le mirvétuximab soravtansine et pendant sept mois après l'arrêt du traitement (voir rubrique Fertilité, grossesse et allaitement).
Excipients à effet notoire
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose, c'est-à-dire qu'il est essentiellement « sans sodium ».
Ce médicament contient 2,11 mg de polysorbate 20 par flacon.
Résumé du profil de sécurité
Les effets indésirables les plus fréquents rapportés avec le mirvétuximab soravtansine étaient : vision trouble (43 %), nausée (41 %), diarrhée (39 %), fatigue (35 %), douleur abdominale (30 %), kératopathie (29 %), sécheresse oculaire (27 %), constipation (26 %), vomissements (23 %), appétit diminué (22 %), neuropathie périphérique (20 %), céphalée (19 %), asthénie (18 %), aspartate aminotransférase - ASAT augmentée (16 %) et arthralgie (16 %).
Les effets indésirables graves les plus fréquemment rapportés étaient : pneumopathie inflammatoire (4 %), occlusion de l'intestin grêle (3 %), occlusion intestinale (3 %), épanchement pleural (2 %), douleur abdominale (2 %), déshydratation (1 %), constipation (1 %), nausée (1 %), ascite (1 %) et thrombopénie (< 1 %).
Les effets indésirables ayant le plus fréquemment entraîné une réduction de dose ou un report du traitement étaient : vision trouble (17 %), kératopathie (10 %), sécheresse oculaire (5 %), neutropénie (5 %), kératite (4 %), cataracte (3 %), baisse de l'acuité visuelle (3 %), thrombopénie (3 %), neuropathie périphérique (3 %) et pneumopathie inflammatoire (3 %).
Le traitement par mirvétuximab soravtansine a été arrêté définitivement en raison d'effets indésirables chez 12 % des patientes ; il s'agissait leplus fréquemment d'affections gastro-intestinales (4 %), d'affections respiratoires, thoraciques et médiastinales (3 %), d'affections hématologiques et du système lymphatique (1 %), d'affections du système nerveux (1%) et d'affections oculaires (1 %).
Liste tabulée des effets indésirables
La fréquence des effets indésirables est basée sur les données combinées de quatre études cliniques menées chez 682 patientes présentant un cancer épithélial de l'ovaire, des trompes de Fallope ou péritonéal primitif (désignés collectivement par cancer épithélial de l'ovaire [CEO]) traitées par le mirvétuximab soravtansine à la dose de 6 mg/kg de poids idéal ajusté administré une fois toutes les trois semaines. La durée médiane de traitement par mirvétuximab soravtansine était de 19,1 semaines (intervalle : 3 à 132 semaines).
La fréquence des effets indésirables rapportés dans les études cliniques est basée sur la fréquence des événements indésirables, toutes causes confondues, pour lesquels il a été déterminé qu'un lien de causalité entre le médicament et l'événement indésirable était au moins raisonnablement possible, après une évaluation approfondie.
Les fréquences sont définies comme suit : très fréquent (≥1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000). Dans chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre de gravité décroissant, le cas échéant.
Tableau 4 : Liste tabulée des effets indésirables tous grades rapportés chez les patientes traitées par le mirvétuximab soravtansine dans les études cliniques
|
Classe de systèmes d'organes |
Fréquence |
Effets indésirables |
|
Infections et infestations |
Très fréquent |
Infection des voies urinaires |
|
Affections hématologiques et du système lymphatique |
Très fréquent |
Anémie, thrombopénie |
|
Fréquent |
Neutropénie |
|
|
Troubles du métabolisme et de la nutrition |
Très fréquent |
Appétit diminué, hypomagnésémie |
|
Fréquent |
Hypokaliémie, déshydratation |
|
|
Affections psychiatriques |
Fréquent |
Insomnie |
|
Affections du système nerveux |
Très fréquent |
Neuropathie périphérique1, céphalée |
|
Fréquent |
Dysgueusie, sensations vertigineuses |
|
|
Affections oculaires |
Très fréquent |
Kératopathie2, cataracte3, épisode de vision trouble[1], photophobie, douleur oculaire, sécheresse oculaire[2] |
|
Fréquent |
Gêne oculaire[3] |
|
|
Affections vasculaires |
Fréquent |
Hypertension |
|
Affections respiratoires, thoraciques et médiastinales |
Très fréquent |
Pneumopathie inflammatoire7, dyspnée, toux |
|
Affections gastro-intestinales |
Très fréquent |
Diarrhée, douleur abdominale[4], constipation, distension abdominale, vomissement, nausée |
|
Fréquent |
Ascite, reflux gastro-œsophagien, stomatite, dyspepsie |
|
|
Affections hépatobiliaires |
Fréquent |
Hyperbilirubinémie |
|
Affections de la peau et du tissu sous-cutané |
Fréquent |
Prurit |
|
Affections musculosquelettiques et du tissu conjonctif |
Très fréquent |
Arthralgie |
|
Fréquent |
Myalgie, dorsalgie, extrémités douloureuses, contractures musculaires |
|
|
Troubles généraux et anomalies au site d'administration |
Très fréquent |
Fatigue |
|
Fréquent |
Fièvre |
|
|
Investigations |
Très fréquent |
Aspartate aminotransférase augmentée, alanine aminotransférase augmentée |
|
Fréquent |
Phosphatase alcaline sanguine augmentée, gamma-glutamyl transférase augmentée, poids diminué |
|
|
Lésions, intoxications et complications d'interventions |
Fréquent |
Réaction liée à la perfusion/hypersensibilité[5] |
1 Le terme groupé « neuropathie périphérique » inclut : hypoesthésie, neuropathie périphérique, neurotoxicité, paresthésie, neuropathie motrice périphérique, neuropathie sensitivomotrice périphérique, neuropathie périphérique sensitive et polyneuropathie (voir la section Description de certains effets indésirables).
2 Le terme groupé « kératopathie » inclut : kyste de la cornée, dépôts cornéens, trouble cornéen, microkystes épithéliaux cornéens, anomalie de l'épithélium cornéen, érosion de la cornée, opacité cornéenne, pigmentation cornéenne, kératite, kératite interstitielle, kératopathie, déficience en cellules souches limbiques et kératite ponctuée (voir la section Description de certains effets indésirables). 3 Le terme groupé « cataracte » inclut : cataracte, cataracte corticale et cataracte nucléaire (voir la section Description de certains effets indésirables).
Description de certains effets indésirables
Affections oculaires
Des effets indésirables oculaires (termes groupés) sont survenus chez 59 % des patientes présentant un CEO traitées par mirvétuximab soravtansine. Onze pour cent (11 %) des patientes ont présenté des effets indésirables oculaires de grade 3 et moins de 1 % des patientes des événements de grade 4. Les effets indésirables oculaires de grade ≥ 3 les plus fréquents étaient : vision trouble et kératopathie (5 % chacun, termes groupés) et cataracte (4 %).
Le délai médian d'apparition du premier effet indésirable oculaire était de 5,1 semaines (intervalle : 0,1 à 68,6 semaines). Chez les patientes ayant présenté des événements oculaires, 53 % ont obtenu une résolution complète (grade 0) et 38 % une amélioration partielle (définie comme une diminution d'un ou plusieurs grades de sévérité par rapport au grade le plus élevé). Lors du dernier suivi, 0,3 % des patientes (2/682) présentait des effets indésirables oculaires de grade ≥3 (baisse de l'acuité visuelle de grade 3 chez une patiente et cataracte de grade 4 chez une patiente).
Les effets indésirables oculaires ont entraîné des reports du traitement chez 24 % des patientes et des réductions de dose chez 15 % des patientes. Les effets indésirables oculaires ont entraîné l'arrêt définitif du traitement par mirvétuximab soravtansine chez 1 % des patientes.
Pneumopathie inflammatoire
Une pneumopathie inflammatoire (termes groupés) est survenue chez 10 % des patientes présentant un CEO traitées par mirvétuximab soravtansine, dont des événements de grade 3 chez 0,9 % des patientes (6/682) et un événement de grade 4 chez 0,2 % des patientes (1/682). Deux patientes (0,3 %) sont décédées des suites d'une insuffisance respiratoire. Une patiente (0,2 %) est décédée d'une insuffisance respiratoire dans le contexte d'une pneumopathie inflammatoire de grade 1 et de métastases pulmonaires confirmées lors de l'autopsie. Une patiente (0,2 %) est décédée d'une insuffisance respiratoire d'étiologie inconnue sans présence d'une pneumopathie inflammatoire concomitante.
Le délai médian d'apparition de la pneumopathie inflammatoire était de 18,1 semaines (intervalle : 1,6 à 97,0 semaines). La pneumopathie inflammatoire a entraîné des reports du traitement par mirvétuximab soravtansine chez 3 % des patientes, des réductions de dose chez 1 % des patientes et l'arrêt définitif du traitement chez 3 % des patientes.
Neuropathie périphérique
Une neuropathie périphérique (termes groupés) est survenue chez 36 % des patientes présentant un CEO traitées par mirvétuximab soravtansine dans les études cliniques ; la neuropathie périphérique était de grade 3 chez 3 % des patientes.
Le délai médian d'apparition de la neuropathie périphérique était de 5,9 semaines (intervalle : 0,1 à 126,7 semaines). La neuropathie périphérique a entraîné des reports du traitement par mirvétuximab soravtansine chez 2 % des patientes, des réductions de dose chez 4 % des patientes et l'arrêt définitif du traitement chez 0,7 % des patientes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SELECTION DES PATIENTES :
Pour être éligibles au traitement, les patientes doivent avoir un
statut tumoral FRα positif, défini comme un marquage membranaire
d'intensité modérée (2+) et/ou forte (3+) dans au moins 75 % des
cellules tumorales viables, déterminé par immunohistochimie (IHC), à
l'aide d'un dispositif médical de diagnostic in vitro (DIV) marqué CE
dans le but recherché correspondant. Si aucun dispositif médical de DIV
marqué CE n'est disponible, un autre test validé doit être utilisé.
AVANT l'instauration du traitement :
- Vérifier l'absence de grossesse.
- Réaliser un examen ophtalmologique comprenant une évaluation de
l'acuité visuelle et un examen à la lampe à fente, ainsi qu'AVANT
l'administration de la dose suivante si une patiente présente de
nouveaux symptômes oculaires ou aggravés.
AVANT chaque perfusion, administrer une prémédication pour la
prévention des réactions liées à la perfusion, des nausées et
vomissements.
SURVEILLANCE du traitement :
- Toxicité oculaire.
- Signes et symptômes pulmonaires de pneumopathie inflammatoire, tels
qu'une hypoxie, une toux, une dyspnée ou des infiltrats interstitiels à
la radiographie. Les étiologies infectieuses, cancéreuses et les autres
causes de ces symptômes doivent être exclues à l'aide d'investigations
appropriées.
- Signes et symptômes de neuropathie tels que paresthésie, sensations
de fourmillements ou de brûlure, douleur neuropathique, faiblesse
musculaire ou dysesthésie.
INFORMER les patientes :
- D'utiliser des larmes artificielles pendant toute la durée du traitement.
- D'éviter de porter des lentilles de contact pendant le traitement par le mirvétuximab soravtansine.
- De ne pas conduire ni utiliser de machines jusqu'à la disparition
complète des symptômes en cas de troubles visuels, de neuropathie
périphérique, de fatigue ou de sensations vertigineuses pendant le
traitement par mirvétuximab soravtansine.
LES PATIENTES EN AGE DE PROCREER doivent utiliser une contraception
efficace pendant le traitement par le mirvétuximab soravtansine et
pendant sept mois après l'arrêt du traitement.
Elles ne doivent pas allaiter pendant le traitement et pendant un mois après la fin du traitement.
Femmes en âge de procréer/Contraception
Chez les patientes en âge de procréer, l'absence de grossesse doit être vérifiée avant l'instauration du traitement par le mirvétuximab soravtansine.
Les patientes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par le mirvétuximab soravtansine et pendant sept mois après l'arrêt du traitement.
Grossesse
Sur la base de son mécanisme d'action, le mirvétuximab soravtansine lorsqu'il est administré à une patiente enceinte, peut avoir des effets délétères sur l'embryon ou le fœtus car il contient un composé génotoxique (DM4) qui agit sur les cellules en division (voir rubriques Propriétés pharmacodynamiques et Données de sécurité préclinique). Les immunoglobulines humaines G (IgG) sont connues pour traverser la barrière placentaire ; par conséquent, le mirvétuximab soravtansine peut être transmis par la mère au fœtus en développement. Il n'existe pas de données disponibles sur l'utilisation du mirvétuximab soravtansine chez des patientes enceintes permettant d'informer sur un risque associé au médicament. Il n'a pas été effectué d'études de toxicité sur la reproduction ou le développement chez l'animal avec le mirvétuximab soravtansine.
L'administration d'ELAHERE aux femmes enceintes n'est pas recommandée, et les patientes doivent être informées des risques potentiels pour le fœtus si elles deviennent enceintes ou prévoient une grossesse. Les patientes qui découvrent qu'elles sont enceintes doivent contacter immédiatement leur médecin. En cas de grossesse survenant pendant le traitement par ELAHERE ou dans les sept mois suivant la fin du traitement, une surveillance étroite est recommandée.
Allaitement
On ne sait pas si le mirvétuximab soravtansine/ses métabolites sont excrétés dans le lait maternel. Un risque pour le nouveau-né/nourrisson ne peut être exclu car les immunoglobulines humaines G (IgG) sont connues pour passer dans le lait maternel. Les femmes ne doivent pas allaiter pendant le traitement et pendant un mois après la fin du traitement par ELAHERE.
Fertilité
Il n'a pas été effectué d'études sur la fertilité avec le mirvétuximab soravtansine ou le DM4. Il n'existe pas de données concernant l'effet d'ELAHERE sur la fertilité humaine. Cependant, du fait du mécanisme d'action d'ELAHERE, qui entraîne la déstabilisation des microtubules et la mort des cellules en division rapide, des effets du médicament sur la fertilité sont possibles.
Il n'a pas été mené d'études cliniques d'interactions médicamenteuses avec ELAHERE.
Le DM4 est un substrat du CYP3A4. L'administration concomitante d'ELAHERE avec des inhibiteurs puissants du CYP3A4 peut augmenter l'exposition au DM4 non conjugué (voir rubrique Propriétés pharmacocinétiques), ce qui peut majorer le risque d'effets indésirables d'ELAHERE (voir rubrique Effets indésirables).Si l'administration concomitante avec des inhibiteurs puissants du CYP3A4 (par exemple céritinib, clarithromycine, cobicistat, idélalisib, itraconazole, kétoconazole, néfazodone, posaconazole, ritonavir, télithromycine, voriconazole) ne peut être évitée, les patientes doivent être étroitement surveillées afin de détecter l'apparition d'effets indésirables. Les inducteurs puissants du CYP3A4 (par exemple, phénytoïne, rifampicine, carbamazépine) peuvent diminuer l'exposition au DM4 non conjugué.
Le traitement par ELAHERE doit être instauré et surveillé par un médecin expérimenté dans l'utilisation des médicaments anticancéreux.
Sélection des patientes
Pour être éligibles au traitement, les patientes doivent avoir un statut tumoral FRα positif, défini comme un marquage membranaire d'intensité modérée (2+) et/ou forte (3+) dans au moins 75 % des cellules tumorales viables, déterminé par immunohistochimie (IHC), à l'aide d'un dispositif médical de diagnostic in vitro (DIV) marqué CE dans le but recherché correspondant. Si aucun dispositif médical de DIV marqué CE n'est disponible, un autre test validé doit être utilisé.
Posologie
La dose recommandée d'ELAHERE est de 6 mg/kg de poids idéal ajusté (PIA), administrée une fois toutes les trois semaines (cycle de 21 jours) en perfusion intraveineuse jusqu'à la progression de la maladie ou toxicité inacceptable. La posologie en fonction du poids idéal ajusté réduit la variabilité de l'exposition chez les patientes en insuffisance pondérale ou en surpoids.
La dose totale d'ELAHERE est calculée en fonction du PIA de chaque patiente selon la formule suivante :
PIA = poids idéal (PI [kg]) + 0,4 × (poids réel [kg] - PI)
Poids idéal chez la femme [kg] = 0,9 × taille [cm] - 92
Pour une patiente d'une taille de 165 cm et pesant 80 kg
|
Calculer d'abord le poids idéal : |
PI = 0,9 × 165 - 92 = 56,5 kg |
|
Puis calculer le poids idéal ajusté : |
PIA = 56,5 + 0,4 × (80 - 56,5) = 65,9 kg |
Prémédication
Prémédication pour la prévention des réactions liées à la perfusion (RLP), des nausées et vomissements
Avant chaque perfusion d'ELAHERE, administrer les prémédications indiquées dans le tableau 1 afin de réduire l'incidence et la sévérité des RLP, des nausées et vomissements.
Tableau 1: Prémédication avant chaque perfusion d'ELAHERE
|
Prémédication |
Voie d'administration |
Exemples (ou équivalents) |
Moment d'administration avant la perfusion d'ELAHERE |
|
Corticoïde |
Intraveineuse |
Dexaméthasone 10 mg |
Au moins 30 minutes avant la perfusion |
|
Antihistaminique |
Orale ou intraveineuse |
Diphénhydramine 25 mg à 50 mg |
|
|
Antipyrétique |
Orale ou intraveineuse |
Paracétamol 325 mg à 650 mg |
|
|
Antiémétique |
Orale ou intraveineuse |
Antagoniste des récepteurs 5HT3 de la sérotonine ou alternatives appropriées |
Avant chaque perfusion et après l'administration des autres prémédications |
Chez les patientes qui présentent des nausées et/ou vomissements, des antiémétiques supplémentaires peuvent être envisagés par la suite, selon les besoins.
Chez les patientes présentant une RLP de grade ≥ 2, une prémédication supplémentaire par dexaméthasone 8 mg deux fois par jour (ou équivalent) la veille de l'administration d'ELAHERE doit être envisagée.
Examen ophtalmologique et prémédication
Examen ophtalmologique : un examen ophtalmologique comprenant une évaluation de l'acuité visuelle et un examen à la lampe à fente doit être réalisé avant l'instauration du traitement par ELAHERE et avant l'administration de la dose suivante si une patiente présente de nouveaux symptômes oculaires ou aggravés. Chez les patientes présentant des effets indésirables oculaires de grade ≥ 2, des examens ophtalmologiques supplémentaires doivent être effectués au moins tous les deux cycles et en fonction du tableau clinique jusqu'à résolution ou retour au grade initial.
Corticoïdes topiques ophtalmiques : chez les patientes présentant des signes d'effets indésirables cornéens (kératopathie) de grade ≥2 lors de l'examen à la lampe à fente, une prophylaxie secondaire par corticoïdes topiques ophtalmiques est recommandée lors des cycles suivants d'ELAHERE, sauf si l'ophtalmologue établit que les risques de ce traitement sont supérieurs aux bénéfices.
- Il doit être recommandé aux patientes d'instiller un collyre corticoïde le jour de la perfusion et pendant les sept jours suivants de chaque cycle ultérieur d'ELAHERE (voir le tableau 3).
- Les patientes doivent être informées qu'elles doivent attendre au moins 15 minutes après l'administration du corticoïde topique ophtalmique avant d'instiller des larmes artificielles.
La mesure de la pression intraoculaire et un examen à la lampe à fente doivent être effectués à intervalles réguliers pendant le traitement par des corticoïdes topiques ophtalmiques.
Larmes artificielles : il est recommandé d'informer les patientes qu'elles doivent utiliser des larmes artificielles pendant toute la durée du traitement par ELAHERE.
Modifications posologiques
Avant le début de chaque cycle, il doit être conseillé aux patientes de signaler au médecin prescripteur ou professionnel de santé qualifié tout nouveau symptôme ou aggravé.
Chez les patientes qui présentent de nouveaux symptômes oculaires ou aggravés, un examen ophtalmologique doit être réalisé avant l'administration d'ELAHERE. Le médecin prescripteur doit passer en revue le compte rendu de l'examen ophtalmologique avant la perfusion et déterminer la dose d'ELAHERE en fonction de la sévérité des anomalies observées dans l'œil le plus sévèrement atteint.
Les réductions de dose et les modifications posologiques en cas d'effets indésirables sont présentées dans le tableau 2 et le tableau 3. Le schéma posologique doit être maintenu à une fréquence d'administration toutes les trois semaines.
Tableau 2 : Schéma de réduction de dose
|
|
Paliers de dose d'ELAHERE |
|
Dose initiale |
6 mg/kg de PIA |
|
Première réduction de dose |
5 mg/kg de PIA |
|
Deuxième réduction de dose |
4 mg/kg de PIA* |
* Le traitement doit être arrêté définitivement chez les patientes qui ne tolèrent pas la dose de 4 mg/kg de PIA.
Tableau 3 : Modifications posologiques en cas d'effets indésirables
|
Effet indésirable |
Sévérité de l'effet indésirable* |
Modification posologique |
|
Kératite/kératopathie (voir rubriques Mises en garde spéciales et précautions d'emploi et Effets indésirables) |
Kératite/kératopathie superficielle non confluente |
Surveiller |
|
Kératite/kératopathie superficielle confluente, anomalie de l'épithélium cornéen ou diminution de 3 lignes ou plus de la meilleure acuité visuelle corrigée |
Suspendre le traitement jusqu'à revenir au stade de kératite/kératopathie superficielle non confluente ou mieux ou jusqu'à la résolution, puis maintenir le même palier de dose. Envisager une réduction de dose chez les patientes présentant une kératite/kératopathie confluente récidivante malgré un traitement symptomatique optimal ou chez les patientes présentant une toxicité oculaire durant plus de 14 jours. |
|
|
Ulcère cornéen ou opacité stromale ou meilleure acuité visuelle corrigée en vision de loin de 6/60 ou moins |
Suspendre le traitement jusqu'à revenir au stade de kératite/kératopathie superficielle non confluente ou mieux ou jusqu'à la résolution, puis réduire la dose d'un palier. |
|
|
Perforation de la cornée |
Arrêter définitivement le traitement. |
|
|
Pneumopathie inflammatoire (voir rubriques Mises en garde spéciales et précautions d'emploi et Effets indésirables) |
Grade 1 |
Surveiller |
|
Grade 2 |
Suspendre le traitement jusqu'à la régression à un grade ≤ 1, puis maintenir au même palier de dose ou envisager une réduction de dose en cas de pneumopathie inflammatoire récidivante, durant plus de 28 jours, ou à l'appréciation du médecin. |
|
|
Grade 3 ou 4 |
Arrêter définitivement le traitement. |
|
|
Neuropathie périphérique (voir rubriques Mises en garde spéciales et précautions d'emploi et Effets indésirables) |
Grade 2 |
Suspendre le traitement jusqu'à la régression à un grade ≤ 1, puis réduire la dose d'un palier. |
|
Grade 3 ou 4 |
Arrêter définitivement le traitement. |
|
|
Réactions liées à la perfusion/hypersensibilité (voir rubriques Mises en garde spéciales et précautions d'emploi et Effets indésirables) |
Grade 1 |
Maintenir le débit de perfusion. |
|
Grade 2 |
• Interrompre la perfusion et administrer un traitement symptomatique. • Après la résolution des symptômes, reprendre la perfusion à un débit inférieur de 50 % et, si absence d'apparition de nouveaux symptômes, augmenter le débit comme approprié jusqu'à la fin de la perfusion. • La veille de la perfusion lors des cycles suivants, administrer une prémédication supplémentaire par dexaméthasone 8 mg par voie orale deux fois par jour (ou équivalent). |
|
|
Grade 3 ou 4 |
• Arrêter immédiatement la perfusion et administrer un traitement symptomatique. • Les patientes doivent être averties qu'elles doivent recevoir un traitement en urgence et informer immédiatement leur médecin ou infirmier/ère en cas de réapparition de symptômes liés à la perfusion après leur sortie du service. • Arrêter définitivement le traitement. |
|
|
Effets indésirables hématologiques (voir rubrique Effets indésirables). |
Grade 3 ou 4 |
Suspendre le traitement jusqu'à la régression à un grade ≤ 1, puis reprendre le traitement à un palier de dose inférieur. |
|
Autres effets indésirables (voir rubrique Effets indésirables). |
Grade 3 |
Suspendre le traitement jusqu'à la régression à un grade ≤ 1, puis reprendre le traitement à un palier de dose inférieur. |
|
Grade 4 |
Arrêter définitivement le traitement. |
* Sauf indication contraire, les grades sont définis selon les Critères de terminologie communs pour les événements indésirables du National Cancer Institute (NCI CTCAE) version 5.0.
Populations particulières
Population pédiatrique
Il n'existe pas d'utilisation justifiée d'ELAHERE dans la population pédiatrique pour le traitement du cancer épithélial de l'ovaire, des trompes de Fallope ou péritonéal primitif (voir rubrique Propriétés pharmacodynamiques).
Sujets âgés
Aucun ajustement posologique d'ELAHERE n'est recommandé chez les patientes âgées ≥ 65 ans (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénale
Aucun ajustement posologique d'ELAHERE n'est recommandé chez les patientes présentant une insuffisance rénale légère à modérée (clairance de la créatinine [ClCr] de 30 à < 90 mL/min). ELAHERE n'a pas été évalué chez les patientes présentant une insuffisance rénale sévère (ClCr de 15 à < 30 mL/min) ou une insuffisance rénale terminale et la nécessité éventuelle d'un ajustement posologique chez ces patientes ne peut pas être déterminée (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucun ajustement posologique d'ELAHERE n'est recommandé chez les patientes présentant une insuffisance hépatique légère (bilirubine totale ≤ limite supérieure de la normale [LSN] et taux d'aspartate aminotransférase ASAT > LSN ou bilirubine totale > 1 à 1,5 × LSN et ASAT quelle que soit la valeur) (voir rubrique Propriétés pharmacocinétiques).
L'utilisation d'ELAHERE chez les patientes présentant une insuffisance hépatique modérée à sévère (bilirubine totale > 1,5 × LSN avec ASAT quelle que soit la valeur) doit être évitée.
Mode d'administration
ELAHERE doit être administré en perfusion intraveineuse à un débit de 1 mg/min. Si la perfusion est bien tolérée après 30 minutes, le débit peut être augmenté à 3 mg/min. Si la perfusion est bien tolérée après 30 minutes à 3 mg/min, le débit peut être augmenté à 5 mg/min.
Pour les incompatibilités, voir la rubrique Incompatibilités.
ELAHERE doit être dilué avec une solution pour perfusion de glucose à 5 %. Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique Précautions particulières d'élimination et de manipulation.
ELAHERE doit être uniquement administré en perfusion intraveineuse, avec un filtre en ligne en polyéthersulfone (PES) de 0,2 µm ou 0,22 µm (voir les procédures particulières de manipulation et d'élimination à la rubrique Précautions particulières d'élimination et de manipulation).
Précautions à prendre avant la manipulation ou l'administration du médicament
Ce médicament contient un composant cytotoxique, lié de façon covalente à l'anticorps monoclonal (voir les procédures particulières de manipulation et d'élimination à la rubrique Précautions particulières d'élimination et de manipulation).
Durée de conservation :
Flacon non ouvert
5 ans
Solution diluée
Après dilution, la stabilité physicochimique de la solution à une concentration de 1 mg/mL à 2 mg/mL a été démontrée pendant 8 heures à une température comprise entre 15 °C et 25 °C ou pendant 24 heures à une température comprise entre 2 °C et 8 °C suivies de 8 heures entre 15 °C et 25 °C.
D'un point de vue microbiologique, le produit doit être utilisé immédiatement, sauf si la méthode de dilution prévient le risque de contamination microbienne. Dans le cas contraire, les durées et conditions de conservation après dilution relèvent de la responsabilité de l'utilisateur.
Précautions particulières de conservation :
Conserver en position verticale au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
Conserver le flacon dans l'emballage extérieur à l'abri de la lumière.
Pour les conditions de conservation du médicament après dilution, voir la rubrique Durée de conservation.
ELAHERE est incompatible avec les solutions pour perfusion de chlorure de sodium à 9 mg/mL (0,9 %). Ce médicament ne doit pas être mélangé avec d'autres médicaments à l'exception de ceux mentionnés dans la rubrique Précautions particulières d’élimination et de manipulation.
Il n'existe aucun traitement/antidote connu en cas de surdosage de mirvétuximab soravtansine. En cas de surdosage, les patientes doivent être étroitement surveillées pour détecter tout signe ou symptôme d'effets indésirables et un traitement symptomatique approprié doit être instauré.
Classe pharmacothérapeutique : Antinéoplasiques et immunomodulateurs, anticorps monoclonaux et anticorps conjugués, autres anticorps monoclonaux et anticorps conjugués.
Code ATC : L01FX26
Mécanisme d'action
Le mirvétuximab soravtansine est un anticorps conjugué. L'anticorps est une IgG1 produite par génie génétique dirigée contre le récepteur alpha du folate (FRα). La fonction de la partie anticorps est de se lier aux récepteurs FRα exprimés à la surface des cellules ovariennes cancéreuses. Le DM4 est un inhibiteur des microtubules conjugué à l'anticorps via un linker clivable. Lors de sa liaison aux récepteurs FRα, le mirvétuximab soravtansine est internalisé, suivi de la libération intracellulaire de DM4 par clivage protéolytique. Le DM4 déstabilise le réseau de microtubules dans la cellule, ce qui entraîne l'arrêt du cycle cellulaire et la mort cellulaire par apoptose.
Effets pharmacodynamiques
Électrophysiologie cardiaque
À la dose recommandée autorisée, le mirvétuximab soravtansine n'a pas entraîné d'augmentations moyennes supérieures à 10 ms de l'intervalleQTc d'après les résultats d'une analyse de la relation concentration- QTc.
Efficacité et sécurité cliniques
Étude IMGN853-0416 (MIRASOL)
L'efficacité et la sécurité du mirvétuximab soravtansine ont été évaluées dans l'étude IMGN853-0416, une étude de phase III, multicentrique, en ouvert, randomisée, contrôlée contre comparateur actif, à deux bras, menée chez des patientes présentant un cancer épithélial séreux avancé de haut grade de l'ovaire, péritonéal primitif ou des trompes de Fallope, résistant aux sels de platine avec positivité pour les récepteurs FRα (y compris sur des échantillons de tissu tumoral archivé), tel que déterminée par le test FOLR1 (FOLR1-2.1) RxDx (intensité de marquage membranaire modérée (2+) et/ou forte (3+) dans au moins 75 % des cellules tumorales viables par immunohistochimie (IHC)).
La maladie résistante aux sels de platine était définie comme un CEO ayant récidivé dans les six mois suivant la dernière administration de platine.
Les patientes présentant un cancer réfractaire primaire aux sels de platine, ayant un score ECOG ≥2 et les patientes présentant des affections cornéennes évolutives ou chroniques, des pathologies oculaires nécessitant un traitement permanent, une neuropathie périphérique de grade ≥ 2 ou une pneumopathie interstitielle/pneumopathie inflammatoire non infectieuse étaient exclues de l'étude.
Les patientes ont été randomisées selon un ratio 1:1 pour recevoir soit ELAHERE à la dose de 6 mg/kg de PIA (N = 227) au jour 1 de chaque cycle de trois semaines, soit l'une des chimiothérapies ci-dessous (N = 226) choisie par l'investigateur avant la randomisation :
1. Paclitaxel (Pac) 80 mg/m2 administré une fois par semaine au cours d'un cycle de quatre semaines ;
2. Doxorubicine liposomale pégylée (DLP) 40 mg/m2 administrée une fois toutes les quatre semaines ;
3. Topotécan (Topo) 4 mg/m2 administré les jours 1, 8 et 15 toutes les quatre semaines ou pendant cinq jours consécutifs à la dose de 1,25 mg/m2 des jours 1 à 5 de chaque cycle de 21 jours.
La randomisation était stratifiée sur le nombre de lignes de traitement antérieures (1 versus 2 versus 3) et sur la chimiothérapie choisie par l'investigateur (Pac versus DLP versus Topo). Le traitement était administré jusqu'à progression de la maladie, décès, retrait du consentement ou survenue d'une toxicité inacceptable.
Le critère principal d'évaluation de l'efficacité était la survie sans progression (SSP) évaluée par l'investigateur selon les critères RECIST 1.1. Le taux de réponse objective (TRO) et la survie globale (SG) étaient les principaux critères secondaires d'évaluation de l'efficacité.
Au total, 453 patientes ont été randomisées. L'âge médian était de 63 ans (intervalle : 29 à 88 ans) et les patientes étaient principalement caucasiennes (66 % ; asiatiques : 12 %). La majorité des patientes (80 %) présentaient un cancer de l'ovaire d'origine épithéliale ; 11 % un cancer des trompes de Fallope ; 8 % un cancer péritonéal primitif ; tous les cancers (100 %) étaient des carcinomes séreux de haut grade. La moitié des patientes environ (47 %) avaient reçu préalablement trois lignes de traitement systémique, 39 % avaient reçu deux lignes de traitement antérieures et 14 % une ligne de traitement antérieure. La majorité des patientes avaient reçu un traitement antérieur par un inhibiteur de la poly(ADP-ribose) polymérase (PARP) (55 %) ou par bevacizumab (62 %). L'intervalle sans sels de platine après la ligne de traitement la plus récente était ≤ 3 mois chez 41 % des patientes, et de 3 à 6 mois chez 58 % des patientes. L'indice de performance ECOG était de 0 chez 55 % des patientes et de 1 chez 44 % des patientes.
L'analyse principale a démontré une amélioration statistiquement significative de la SSP et de la SG chez les patientes randomisées recevant ELAHERE par rapport aux patientes recevant la chimiothérapie choisie par l'investigateur.
Une synthèse des résultats d'efficacité de l'étude IMGN853-0416 (MIRASOL) est présentée dans le tableau 5.
Tableau 5 : Résultats d'efficacité de l'étude IMGN853-0416
|
Paramètre d'efficacité |
ELAHERE N = 227 |
Chimiothérapie choisie par l'investigateur N = 226 |
|
Survie sans progression (SSP) selon l'évaluation par l'investigateur |
||
|
Nombre d'événements (%) |
176 (77,5) |
166 (73,5) |
|
Médiane, mois (IC à 95 %) |
5,62 (4,34 ; 5,95) |
3,98 (2,86 ; 4,47) |
|
Hazard ratio (IC à 95 %) |
0,65 (0,521 ; 0,808) |
|
|
Valeur de p |
< 0,0001 |
|
|
Survie globale (SG) |
||
|
Nombre d'événements (%) |
90 (39,6) |
114 (50,4) |
|
Médiane, mois (IC à 95 %) |
16,46 (14,46 ; 24,57) |
12,75 (10,91 ; 14,36) |
|
Hazard ratio (IC à 95 %) |
0,67 (0,504 ; 0,885) |
|
|
Valeur de p |
0,0046* |
|
Date de recueil des données fixée au 6 mars 2023.
*:
Limite d'efficacité prédéfinie = 0,01313, test bilatéral (avec
ajustement pour le nombre de décès observés, 204).
Les courbes de Kaplan Meier de la SSP évaluée par l'investigateur (durée médiane de suivi de 11,2 mois) et de la SG (durée médiane de suivi de 13,1 mois) sont présentées dans la Figure 1 et la Figure 2.
Figure 1 : Courbe de Kaplan Meier de la survie sans progression dans l'étude MIRASOL, par bras de traitement (population en intention de traiter)
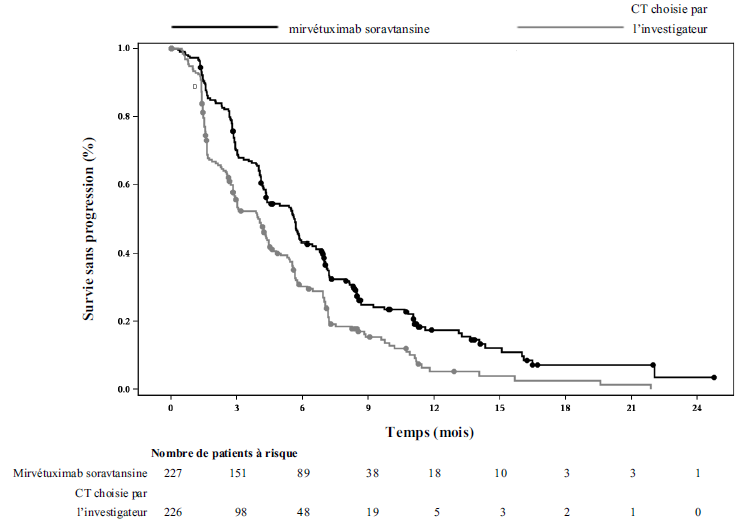
Figure 2 : Courbe de Kaplan Meier de la survie globale dans l'étude MIRASOL, par bras de traitement (population en intention de traiter)
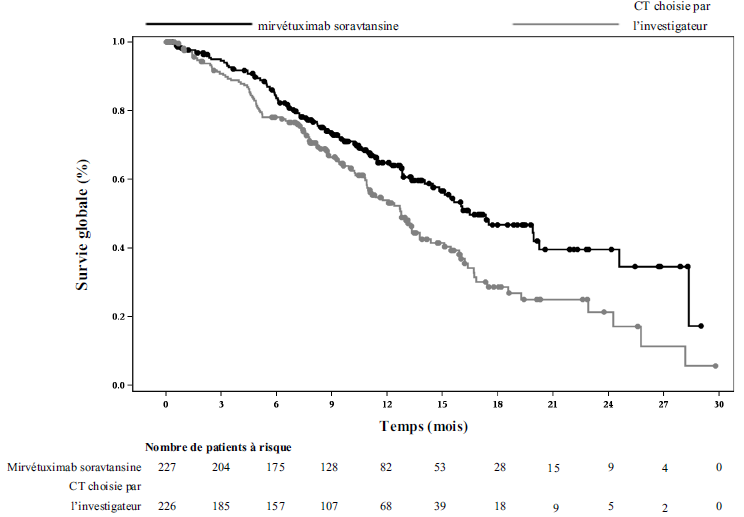
Lors d'une analyse descriptive supplémentaire avec une durée médiane de suivi de 20,3 mois, les résultats de SG concordaient avec ceux observés dans l'analyse principale.
Immunogénicité
Des anticorps anti-médicament (AAM) ont été détectés fréquemment. Il n'a pas été observé de signes d'un effet des AAM sur la pharmacocinétique, l'efficacité ou la sécurité ; cependant, les données sont encore limitées.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec ELAHERE dans tous les sous-groupes de la population pédiatrique dans le traitement du carcinome de l'ovaire, des trompes de Fallope et du péritonéal (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Sauf indication contraire, la pharmacocinétique a été caractérisée après administration de mirvétuximab soravtansine aux doses de 0,161 mg/kg à 8,71 mg/kg de PIA (soit 0,0268 fois à 1,45 fois la dose recommandée autorisée de 6 mg/kg de PIA).
Le tableau 6 présente une synthèse des paramètres d'exposition au mirvétuximab soravtansine, au DM4 non conjugué et à son métabolite S-méthyl DM4 après le premier cycle (3 semaines) de mirvétuximab soravtansine administré à la dose de 6 mg/kg. Les concentrations plasmatiques maximales du mirvétuximab soravtansine ont été observées à peu près à la fin de la perfusion intraveineuse, tandis que les concentrations maximales du DM4 non conjugué ont été observées le deuxième jour suivant l'administration de mirvétuximab soravtansine, et que les concentrations maximales du S-méthyl DM4 ont été observées environ trois jours après la perfusion. Les concentrations à l'état d'équilibre du mirvétuximab soravtansine, du DM4 et du S-méthyl DM4 étaient atteintes après un cycle de traitement. Après administration répétée de mirvétuximab soravtansine, l'accumulation du mirvétuximab soravtansine, du DM4 et du S-méthyl DM4 était minime.
Tableau 6 : Paramètres d'exposition au mirvétuximab soravtansine, au DM4 non conjugué et au S-méthyl-DM4 après le premier cycle de mirvétuximab soravtansine à la dose de 6 mg/kg
|
|
Mirvétuximab soravtansine Moyenne (± ET) |
DM4 non conjugué Moyenne (± ET) |
S-méthyl DM4 Moyenne (± ET) |
|
Cmax |
137,3 (± 62,3) µg/mL |
4,11 (± 2,29) ng/mL |
6,98 (± 6,79) ng/mL |
|
ASCtau |
20,65 (± 6,84) h*mg/mL |
530 (± 245) h*ng/mL |
1 848 (± 1 585) h*ng/mL |
Cmax = concentration maximale, ASCtau = aire sous la courbe de la concentration en fonction du temps sur l'intervalle posologique (21 jours).
Absorption
Le mirvétuximab soravtansine est administré en perfusion intraveineuse. Il n'a pas été mené d'études avec d'autres voies d'administration.
Distribution
Le volume de distribution moyen (± écart type [ET]) du mirvétuximab soravtansine à l'état d'équilibre était de 2,63 (± 2,98) litres. In vitro, le taux de liaison du DM4 et du S-méthyl DM4 aux protéines plasmatiques humaines était supérieur à 99 %.
Biotransformation
La partie anticorps monoclonal du mirvétuximab soravtansine devrait être métabolisée en petits peptides par des voies cataboliques. Le métabolisme du DM4 non conjugué et du S-méthyl DM4 fait intervenir le CYP3A4. Dans le plasma humain, le DM4 et le S-méthyl DM4 ont été identifiés comme les principaux métabolites en circulation, et représentaient environ 0,4 % et 1,4 % respectivement des ASC du mirvétuximab soravtansine.
Élimination
La clairance plasmatique totale moyenne (± ET) du mirvétuximab soravtansine était de 18,9 (± 9,8) mL/heure. La demi-vie d'élimination terminale moyenne du mirvétuximab soravtansine après administration de la première dose était de 4,9 jours. Pour le DM4 non conjugué, la clairance plasmatique totale moyenne (± ET) était de 14,5 (± 4,5) mL/heure et la demi-vie d'élimination terminale moyenne était de 2,8 jours. Pour le S-méthyl DM4, la clairance plasmatique totale moyenne (± ET) était de 5,3 (± 3,4) L/heure et la demi-vie d'élimination terminale moyenne était de 5,1 jours. Les études in vitro et précliniques in vivo indiquent que le DM4 et le S-méthyl DM4 sont métabolisés principalement par le CYP3A4 et éliminés par excrétion biliaire dans les fèces.
Populations particulières
Il n'a pas été observé de différences cliniquement significatives de la pharmacocinétique du mirvétuximab soravtansine en fonction de l'âge (32 à 89 ans), du groupe ethnique (caucasien, noir ou asiatique), du poids (36 à 136 kg) ou de la présence d'une insuffisance hépatique légère (bilirubine totale ≤ LSN et ASAT quelle que soit la valeur > LSN ou bilirubine totale < 1 à 1,5 × LSN et ASAT quelle que soit la valeur) ou d'une insuffisance rénale légère à modérée (ClCr≥ 30 et < 90 mL/min). La pharmacocinétique du mirvétuximab soravtansine chez les patientes présentant une insuffisance hépatique modérée à sévère (bilirubine totale > 1,5 × LSN avec ASAT quelle que soit la valeur) ou une insuffisance rénale sévère (ClCr de 15 à 30 mL/min) n'a pas été établie.
Études d'interactions
Études in vitro
Enzymes du cytochrome P450 (CYP) : le DM4 non conjugué exerce un effet inhibiteur dépendant du temps sur le CYP3A4. Le DM4 non conjugué et le S-méthyl DM4 ne sont pas des inhibiteurs directs des CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 et CYP3A. Le DM4 et le S-méthyl DM4 ne sont pas des inducteurs des CYP1A2, CYP2B6 et CYP3A4.
Transporteurs : le DM4 non conjugué et le S-méthyl DM4 sont des substrats de la P-gp, mais ne sont pas des inhibiteurs de ce transporteur.
ELAHERE a une influence modérée sur l'aptitude à conduire des véhicules et à utiliser des machines. Les patientes doivent être informées qu'en cas de troubles visuels, de neuropathie périphérique, de fatigue ou de sensations vertigineuses pendant le traitement par mirvétuximab soravtansine, elles ne doivent pas conduire ni utiliser de machines jusqu'à la disparition complète des symptômes.
Les organes cibles identifiés après administration d'une dose unique de mirvétuximab soravtansine chez le singe cynomolgus étaient limités à la peau, à la moelle osseuse et au tissu lymphoïde (hypoplasie). Après administrations répétées chez le singe cynomolgus et le lapin hollandais, il a également été observé des anomalies oculaires incluant des microkystes de la cornée, une pigmentation cornéenne, une diminution et une dégénérescence/nécrose de l'épithélium cornéen. Ces anomalies étaient dépendantes de l'intensité de la dose (dose et schéma posologique), une incidence globale plus faible de ces anomalies et une régression étant observées avec le schéma d'administration toutes les trois semaines (le schéma d'administration en clinique).
Il n'a pas été effectué d'études de cancérogenèse avec le mirvétuximab soravtansine ou le DM4.
Le DM4 et le S-méthyl DM4 n'étaient pas mutagènes dans l'essai de mutation réverse sur bactéries (test d'Ames). Le DM4 et le S-méthyl DM4 ont provoqué la formation de micronoyaux dans les érythrocytes polychromatiques.
Aucune étude de toxicité sur la reproduction ou le développement n'a été conduitechez l'animal avec le mirvétuximab soravtansine.
Aucune étude de fertilité chez l'animaln'a été menée avec le mirvétuximab soravtansine ou le DM4. Il n'existe pas de données concernant l'effet d'ELAHERE sur la fertilité humaine. Cependant, du fait du mécanisme d'action d'ELAHERE qui entraîne la déstabilisation des microtubules et la mort des cellules en division rapide, des effets du médicament sur la fertilité sont possibles.
ELAHERE est un médicament cytotoxique. Respecter les procédures particulières pour la manipulation et l'élimination.
Préparation
- Calculer la dose (en mg) (en fonction du poids idéal ajusté [PIA] de la patiente), le volume total (en mL) de solution requis et le nombre de flacons d'ELAHERE nécessaire (voir rubrique Posologie et mode d'administration). Plusieurs flacons peuvent être nécessaires pour obtenir une dose complète.
- Sortir les flacons d'ELAHERE du réfrigérateur et les laisser réchauffer à température ambiante.
- Lorsque la solution et le flacon le permettent, les médicaments à usage parentéral doivent être inspectés avant l'administration pour vérifier l'absence de particules et de coloration anormale. ELAHERE est une solution incolore limpide à légèrement opalescente.
- Le médicament ne doit pas être utilisé si la solution est trouble, présente une coloration anormale ou contient des particules étrangères.
- Avant de prélever le volume de la dose calculée d'ELAHERE pour la dilution ultérieure, faire tourner doucement et examiner chaque flacon. Ne pas agiter le flacon.
- Dans des conditions aseptisées, prélever le volume de la dose calculée d'ELAHERE pour la dilution ultérieure.
- ELAHERE ne contient pas de conservateurs et est à usage unique. Éliminer toute solution non utilisée restant dans le flacon.
Dilution
- Avant l'administration, ELAHERE doit être dilué avec une solution de glucose à 5 % pour obtenir une concentration finale de 1 mg/mL à 2 mg/mL.
- ELAHERE est incompatible avec les solutions pour perfusion de chlorure de sodium à 9 mg/mL (0,9 %). ELAHERE ne doit pas être mélangé avec d'autres médicaments ni avec des solutés intraveineux.
- Déterminer le volume de solution de glucose à 5 % nécessaire pour obtenir la concentration finale de la substance active diluée. Soit retirer le volume de solution de glucose à 5 % en excès d'une poche à perfusion intraveineuse préremplie ou ajouter le volume calculé de solution de glucose à 5 % dans une poche à perfusion intraveineuse vide stérile. Puis ajouter le volume de la dose calculée d'ELAHERE dans la poche à perfusion.
- Mélanger doucement la solution diluée en retournant lentement la poche plusieurs fois pour garantir un mélange uniforme. Ne pas secouer ni agiter.
- Si la solution pour perfusion diluée n'est pas utilisée immédiatement, elle doit être conservée conformément à la rubrique Durée de conservation. Si la poche à perfusion est conservée au réfrigérateur, la laisser atteindre la température ambiante avant l'administration. Après réfrigération, la solution pour perfusion diluée doit être administrée dans les huit heures (en incluant la durée de perfusion).
- Ne pas congeler la solution pour perfusion préparée.
Administration
- Avant l'administration, examiner la poche à perfusion intraveineuse d'ELAHERE pour vérifier l'absence de particules et de coloration anormale.
- Administrer les prémédications avant la perfusion d'ELAHERE (voir rubrique Posologie et mode d'administration).
- ELAHERE ne doit être administré qu'en perfusion intraveineuse, avec un filtre en ligne en polyéthersulfone (PES) de 0,2 µm ou 0,22 µm. Ne pas utiliser d'autres types de membrane.
- L'utilisation de perfuseurs contenant du phtalate de di-2-éthylhexyle (DEHP) doit être évitée.
- Administrer la dose initiale en perfusion intraveineuse à un débit de 1 mg/min. Si la perfusion est bien tolérée après 30 minutes à 1 mg/min, le débit peut être augmenté à 3 mg/min. Si la perfusion est bien tolérée après 30 minutes à 3 mg/min, le débit peut être augmenté à 5 mg/min.
- Si la patiente n'a pas présenté de réactions liées à la perfusion lors de l'administration de la dose précédente, les perfusions suivantes doivent débuter au débit maximal toléré, qui peut être augmenté jusqu'à un débit maximal de 5 mg/min, en fonction de la tolérance.
- Après la perfusion, rincer la ligne intraveineuse avec une solution de glucose à 5 % pour garantir la délivrance de la dose complète. Ne pas utiliser d'autres solutés intraveineux pour le rinçage.
Élimination
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription réservée aux médecins compétents en CANCEROLOGIE.
Prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE.
Réservé à l'usage HOSPITALIER.
Solution à diluer pour perfusion (solution à diluer stérile).
Solution incolore limpide à légèrement opalescente
Flacon en verre de type I muni d'un bouchon en caoutchouc butyle et d'une capsule en aluminium avec opercule en polypropylène de couleur bleu roi contenant 20 mL de solution à diluer.
Boîte d'1 flacon.
1 mL de solution à diluer pour perfusion contient 5 mg de mirvétuximab soravtansine. Un flacon contient 100 mg de mirvétuximab soravtansine dans 20 mL.
Le mirvétuximab soravtansine est un conjugué anticorps-médicament (anticorps conjugué) dirigé contre le récepteur alpha du folate (FRα). L'anticorps conjugué est composé d'un anticorps monoclonal de sous type IgG1 anti-FRα, produit selon la technologie de l'ADN recombinant dans des cellules ovariennes de hamster chinois (CHO), et conjugué via un linker clivable (ester d'acide butanoïque, 4-(2-pyridinyldithio)-2-sulfo-1-(2,5-dioxo-1-pyrrolidinyl)) à un maytansinoïde DM4, un agent anti-tubuline. Le mirvétuximab soravtansine contient en moyenne 3,4 molécules du cytotoxique DM4 liées à l'anticorps anti-FRα.
Excipient(s) à effet notoire
Ce médicament contient 2,11 mg de polysorbate 20 par flacon.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Acide acétique glacial (E260)
Acétate de sodium (E262)
Saccharose
Polysorbate 20 (E432)
Eau pour préparations injectables